Predicting FDA Approval of Small Molecule Drugs¶
Background¶
The drug development process is a high-risk, long-term, and expensive endeavor. On average, tens of thousands of candidates enter the drug discovery pipeline for every one that becomes FDA-approved. In addition to this high attrition, the process takes 10 years and is estimated to cost $1.4 billion. Machine Learning could help reduce the risk, thereby lowering costs and expediting the process.
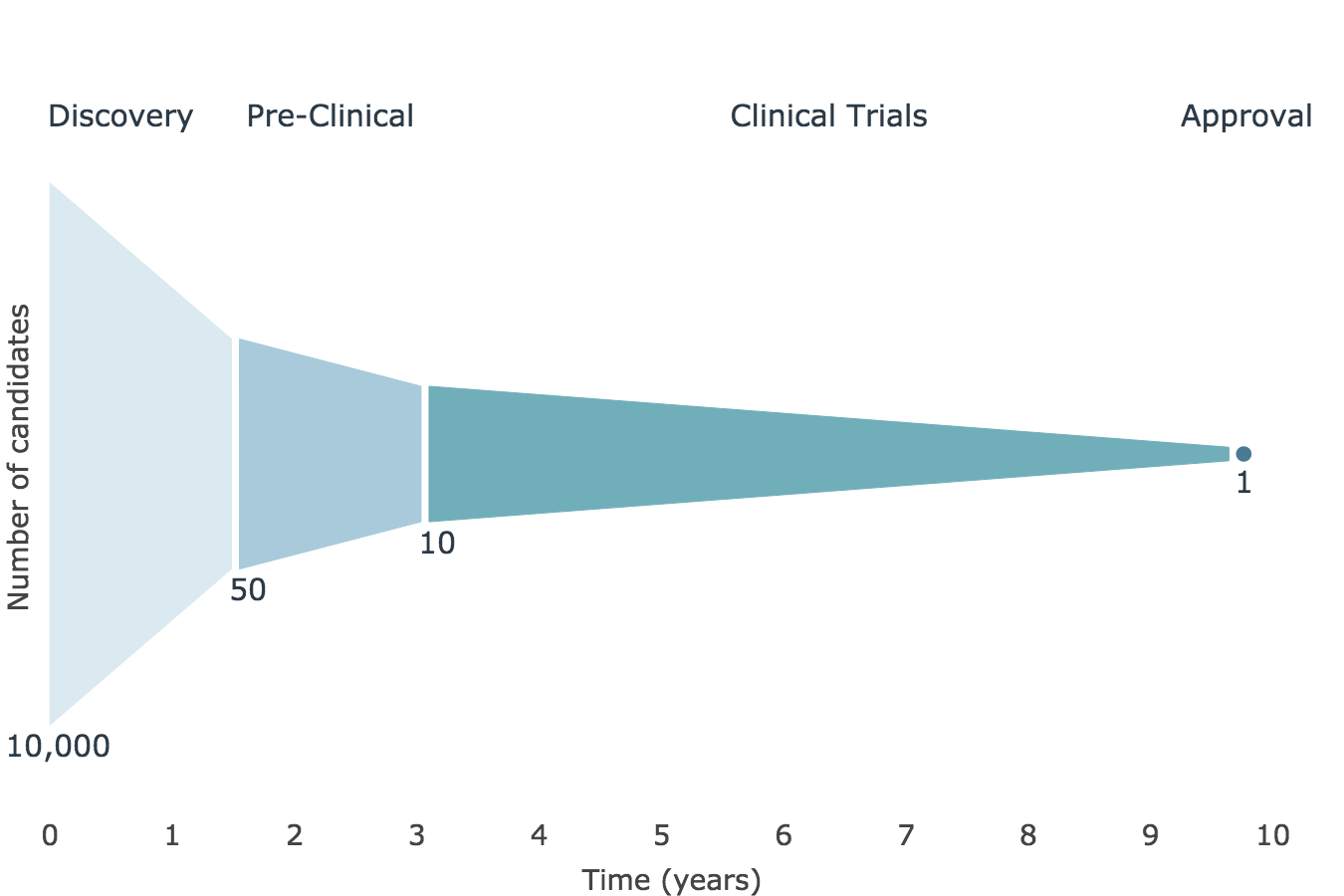
Data¶
My data source was ChEMBL, a chemical database curated by the European Molecular Biology Laboratory. It was hosted on Google BigQuery and was also available for download as a PostgreSQL database. It contains information on over 1 million drug-like molecules in 77 tables. I decided to focus on small molecules. After cleaning the data and excluding any entries from the past 10 years, I was left with 432,000 entries. My dataset was highly imbalanced, as only 0.4% of these small molecules attained FDA approval.
Methods¶
SQL¶
I used the Google BigQuery Python Client Library bigquery and the helper class bq_helper to query the ChEMBL database. I performed a series of SQL queries to extract data from SQL tables into pandas dataframe.
Feature Selection & Engineering¶
The ‘COMPOUND_PROPERTIES’ table contained chemical properties of the molecules. These were mostly numerical features and were thus an obvious choice to include. Other tables contained more categorical variables with large numbers of levels. For assays (395 levels) and targets (9134 levels), I took their assigned parent class. For target organisms (2427 levels), I created a Boolean variable: human or not.
I tallied the number of target organisms by molecule. The number of targets and activities were highly correlated, so I merged these into one feature by taking their average. These were also correlated with the number of assays, so I computed the ratio of this average with the number of assays.
A given small molecule might have multiple activity entries -- in other words, it could have one or more targets and assays associated with it. I thus couldn't simply use OneHotEncode on these features, so I used CountVectorize to encode this information instead.
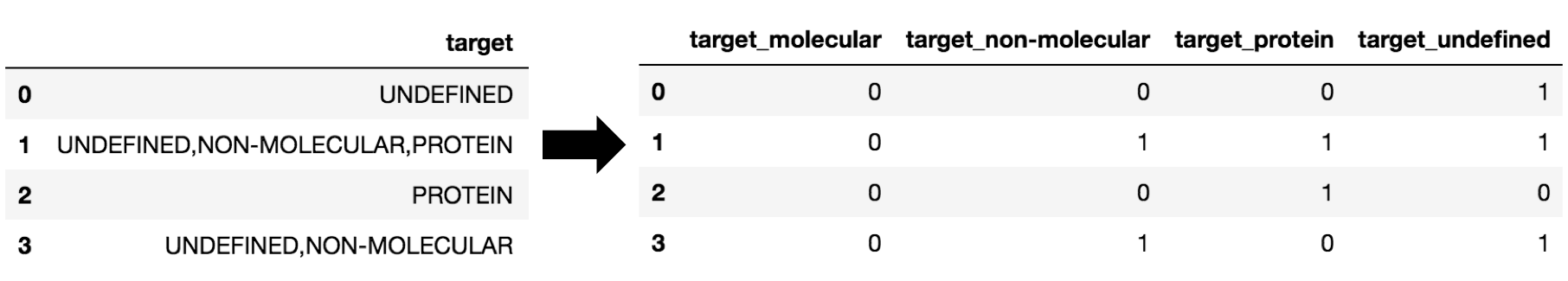
A list of final features is given in my GitHub repository.
Pre-Processing¶
Before sending my data into classifier algorithms, I had to preprocess it as follows:
- Impute missing values
- Scale numerical variables (for Logistic Regression)
- Count-Vectorize the categorical variables for which entries can take on >1 level (assay, target)
- One-Hot-Encode the other categorical variables
- Engineer features
Because of the diversity of my feature datatypes, I decided to make use of scikit-learn’s Pipelines, using the ColumnTransformer, FunctionTransformer, and FeatureUnion functions. I also created custom TransformerMixin Classes to handle some of the preprocessing tasks. I will write a future blog post about how I did this. It took a lot of time to develop, but once I had the code working, processing my test dataset was a breeze.
Modeling¶
I evaluated Logistic Regression, Random Forest, Bagged Decision Trees, and “vanilla” Gradient Boosting algorithms. These were all out-performed by CatBoost, an algorithm based on gradient boosting decision trees, which is especially fast and adept at handling categorical variables. I also investigated various methods of over- and under-sampling to address the imbalance in my dataset including SMOTE, ADASYN, Random Undersampling, Edited Nearest Neighbors, Near Miss versions 1 & 3, SMOTE-EEN, SMOTE-Tomek, and assigning class weights. None improved the performance of my models.
I then tuned the hyperparameters of CatBoost using BayesSearchCV. I may write a separate blog post on how I did this, as CatBoost is a newer algorithm and there are not a lot of examples out there on using it. Here, I report my final hyperparameters; while these details may border on boring, I feel it's important to share them in an effort to make my work reproducible:
| Hyperparameter | Tuned Value | Default |
|---|---|---|
| Hyperparameter | Tuned Value | Default |
| Tree Depth | 7 | 6 |
| Learning rate | 0.084 | 0.03 |
| L2 Regularization | 0.892 | 3 |
| Random Strength | 16.7 | 1 |
| Bagging Temperature | 0.55 | None |
Results¶
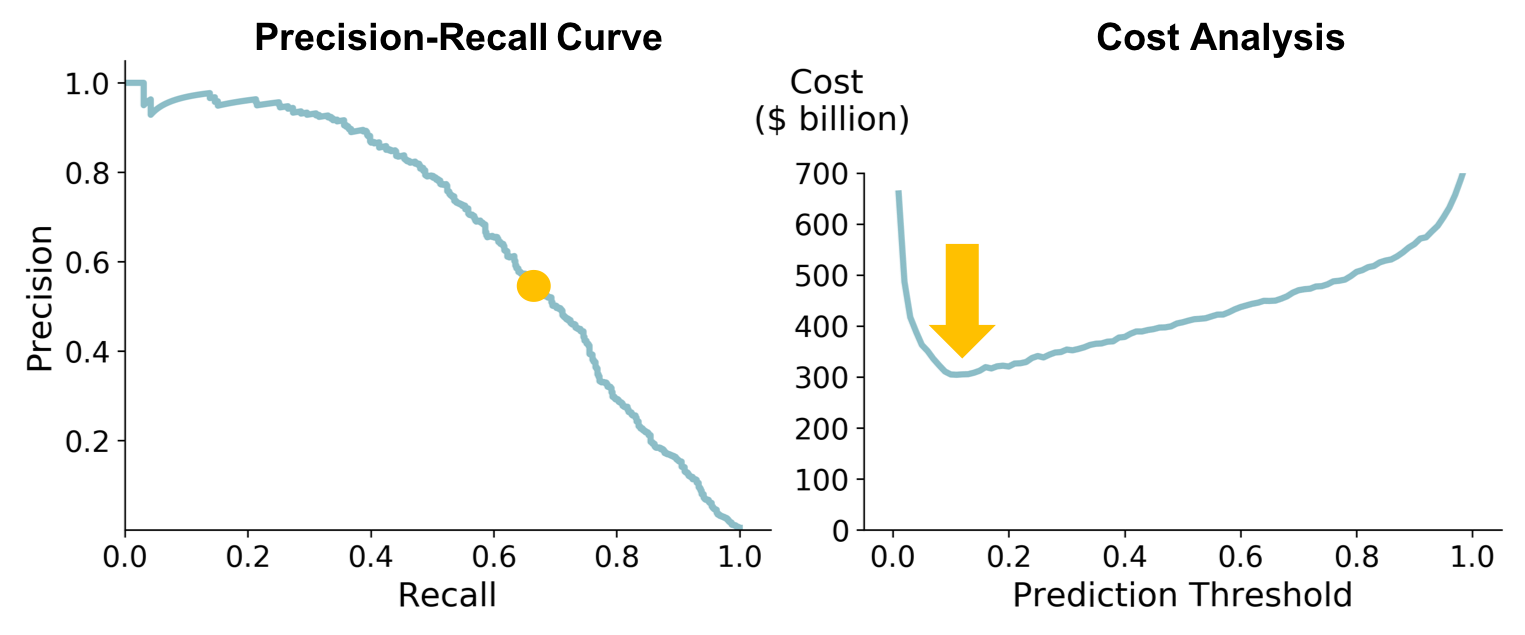
The F1-score for my training and test (hold-out) sets is given in the table below; the Precision-Recall curve for the test set is shown below. The training score is from the BayesSearchCV hyper-tuning, which used 5-fold stratified cross-validation.
| Dataset | F1-score |
|---|---|
| Dataset | F1-score |
| Train | 0.575 |
| Test | 0.593 |
The test set actually had a higher F1-score than the training set, indicating that the model is not overfitting.
I used a cost function to determine the best threshold to use. Based on industry numbers, I estimated the cost of a false-positive to be $\$$200 million (the cost to develop a drug) and the cost of a false-negative to be $\$$1 billion (lost revenue). Given the relatively high cost of a false-negative, the cost function is optimized at a low threshold (~ 0.15). The corresponding point on the Precision-Recall curve is shown below.
The most logical way in which to apply my model to the drug discovery process is in the early phases. This is essentially where the model was trained: on thousands of compounds, most of which never see the light of day. Using the model here, it could reduce the number of compounds under consideration at this stage, thereby expediting the screening process and reducing testing costs. It would furthermore increase our confidence in the molecules sent forward in the pipeline to Pre-Clinical testing. I estimate this could save 6-12 months and up to $\$$100 million.
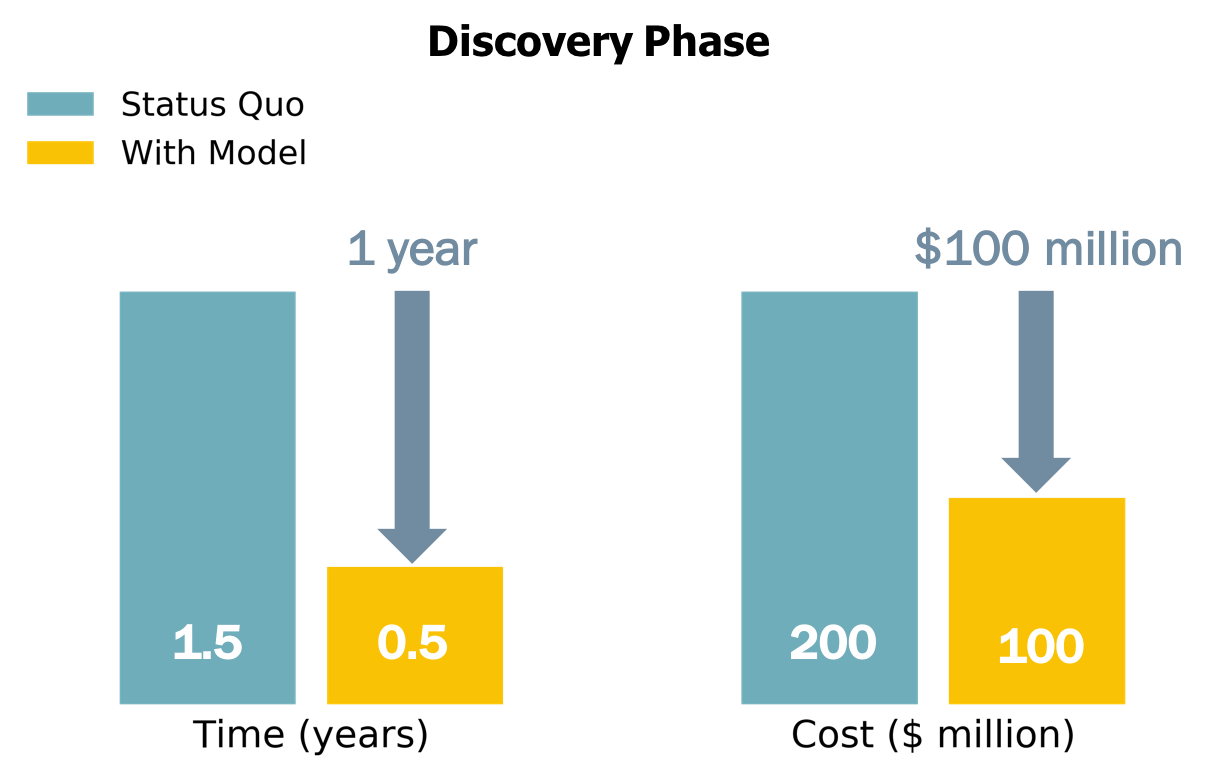
1 year and $\$$100 million might seem paltry in the face of 10 years and $\$$1.4 billion but it’s a good first step towards reducing the time, expesne, and risk involved in bringing a new drug to market.
Future Work¶
Include more bioactivity data I had grand visisons of ranking molecules according to their actvitiy; using NLP (natural language processing) to extract information from the assay descriptions; even creating a "Recommender System" for small molecules & their targets (think of the small molecules as the 'users' and the targets as the 'items')...alas, time is finite.
Expand to Biologics Biologics are protein or protein fragment drugs. They are vastly more complex molecules than the small ones I examnined here, making their analysis more complicated. They are also where much of the innovation in drug discovery is occuring, so they are a worthwhile challenge to take on.